Epidermolysis bullosa (EB)
Quick Search
- Summary
- Synonyms and Classifications
- Symptoms
- Disability Impacts
- Cause and Inheritance
- Diagnosis
- Treatment
- Clinical Care Team
- Clinical Care Guidelines
- Emergency Management
- Research
- Rare Disease Organisation(s)
- Lived Experience
- Support Services and Resources
- Mental Health
- Other Information
- Useful Links for Healthcare Professionals
Summary
Epidermolysis bullosa (EB) is a group of genetic skin conditions in which the skin is very fragile and can injure easily.1-3 This can occur even from a slight touch or friction, resulting in blistering and peeling of the skin. Symptoms can range from mild with slight blisters to severe with painful blisters requiring regular dressings as well as with other complications.2,3
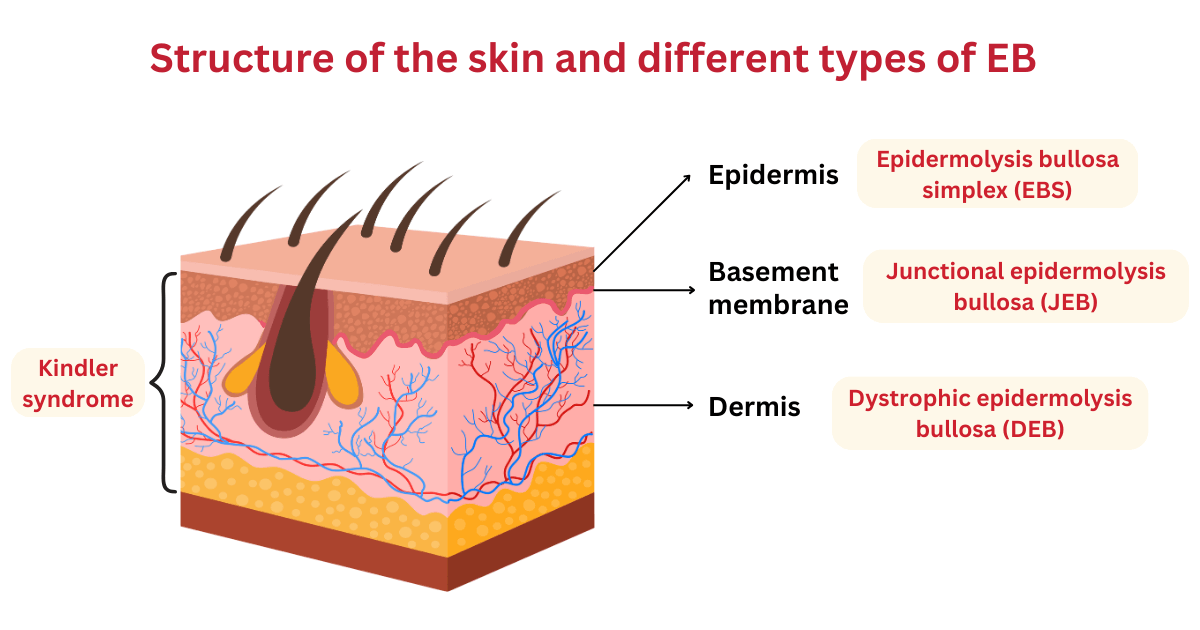
The skin consists of two main layers – the epidermis (top layer) and dermis (inner layer), which are separated by the basement membrane. There are four main types of EB, which are characterised by the layer/s of skin that are affected:2,4
- Epidermolysis bullosa simplex (EBS) [ORPHA:304] – affects the top layer of the skin (epidermis);most common form of EB
- Junctional epidermolysis bullosa (JEB) [ORPHA:305] – blistering occurs in the basement membrane, which is between the top layer and the inner layer of the skin
- Dystrophic epidermolysis bullosa (DEB) [ORPHA:303] – blistering occurs in the inner layer of the skin (dermis); scarring often occurs
- Kindler syndrome (mixed EB) [ORPHA:2908] – affects multiple layers of the skin; skin is sensitive to sun (photosensitive) and can develop poikiloderma, which includes changes in pigmentation, thinning of the skin (atrophy) and visible blood vessels (telangiectasia)
EB usually presents at birth or early on in life.1,2 It is a heritable condition (can be passed on from parents to children).1 EB is not contagious, and the symptoms of EB cannot be passed on through skin contact.2
There is a separate condition called Epidermolysis bullosa acquisita [ORPHA:46487], which is an autoimmune condition and is not genetic. Treatment for Epidermolysis bullosa acquisita may differ from EB.
Synonyms and Classifications
Synonyms: Acantholysis bullosa; inherited epidermolysis bullosa; epidermolysis bullosa hereditaria; hereditary epidermolysis bullosa.
Universal rare disease classifications provide a common language for recording, reporting and monitoring diseases. Please visit the Rare Disease Classifications page for more information about these internationally recognised classifications.
Symptoms
EB is characterised by fragile skin that can injure easily, causing blisters and wounds.1,2 These blisters can occur anywhere on the skin and in the mouth (oral cavity), and in some cases affect the eyes, respiratory tract, digestive (gastrointestinal) system, and genital and urinary (genitourinary) system.3 The wounds may be slow to heal and can become inflamed, infected and at times cause scarring.1 Blisters in the mouth, digestive and genitourinary system can cause difficulty eating, malnutrition, poor growth and constipation.2
The extent of the symptoms varies between the different types of EB and can range from mild to severe.1-3 With the mild cases of EB, there may be only a few blisters affecting mostly the hands and feet with low likelihood of scarring.1,2 In the severe cases, the wounds may be extensive and painful with increased risk of infections.1 This can result in disabilities affecting daily life and complications, some of which can be life-threatening.1,4 Complications of EB depends on the type of EB and include, but may not be limited to, sepsis, anaemia, osteoporosis, kidney disease, respiratory issues, and skin cancer.1,3
Please speak to your medical team to learn more about the symptoms and complications of a specific type or subtype of EB.
Disability Impacts
Rare diseases are often serious and progressive, exhibiting a high degree of symptom complexity, leading to significant disability. Majority of the estimated two million Australians living with a rare disease meet the Australian Government’s definition for disability (in accordance to the Australian Public Service Commission and Australian Bureau of Statistics), and many experience severe and permanent disability impacts. If you or someone you care for is experiencing disability-related impacts from a rare condition, please speak with a health or disability professional for advice. Information about relevant disability support can be found at the RARE Portal’s Disability Support Information page.
Cause and Inheritance
EB is a group of genetic conditions and is caused by disease-causing genetic changes (variants) in specific genes that are involved in skin structure, integrity and repair.3,6 These genetic variants result in fragility of the skin.3
EB can be inherited in either an autosomal dominant or autosomal recessive manner depending on the type of EB.3,6
More information on the relevant inheritance patterns can be found at:
- Centre for Genetics Education: Autosomal dominant inheritance
- Centre for Genetics Education: Autosomal recessive inheritance
If you would like to learn more about the inheritance and impact of a particular type of EB, please ask your doctor for a referral to a genetic counsellor. Genetic counsellors are qualified allied health professionals who can provide information and support regarding genetic conditions and testing. More information about genetic counselling can be found at:
- Information on Genetic Services
- The National and State Services pages underneath the ‘Genetic Counselling’ sections listed
Diagnosis
Diagnosis of EB, including the identification of the particular type of EB, may be made based on detailed family history, clinical examination for characteristic signs of EB and a skin biopsy to identify structural abnormalities in the skin. Genetic testing may be used to confirm the diagnosis.1,3,4
In cases where there is a family history of EB, and the causal gene and its genetic change has been identified, prenatal diagnosis may be an option.1
A differential diagnosis can rule out other conditions with similar symptoms, particularly in the newborns, such as infections, pompholyx, immunobullous disease, porphyria, ichthyoses, palmoplantar keratoderma, Ehlers-Danlos syndromes (EDS), incontinentia pigmenti, Poikiloderma syndromes, Bullous mastocytosis and Epidermolysis bullosa acquisita.3-5
Please speak to your medical team to learn more about the available diagnostic pathways for EB.
Treatment
There is currently no curative treatment for EB. Treatment is targeted at managing symptoms, reduce the development of new blisters and infection, and promote skin healing. Management of EB typically involves a multidisciplinary medical team and may include strategies such as:1-4
- gentle handling to reduce friction or rubbing against the skin
- use of appropriate clothing and footwear, such as loose-fitting clothing
- keeping cool whenever possible
- draining of new blisters in a sterile manner to relieve pain and limit its growth
- treating of wounds and prevention of infections using creams, dressings and antiseptic washes as appropriate
- targeted antibiotic use
- pain management
- management of nutritional deficiencies
- monitoring for complications, including cancer
Individuals with an EB diagnosis may be eligible to access approved dressings through the government-subsidised National Epidermolysis Bullosa Dressing Scheme. An Australian Government Department of Health and Aged Care: National Epidermolysis Bullosa Dressing Scheme administrator is available for contact to help with the costs of epidermolysis bullosa (EB) treatment.
Please speak to your medical team to learn more about the possible treatment or management options for your condition. Treatment will depend on an individual’s specific condition and symptoms. It is also important to stay connected to your medical team so that you can be made aware of any upcoming clinical trial opportunities. For many rare diseases, treatment options may be limited. Participation in a clinical trial may provide access to new or emerging therapies.
Clinical Care Team
Healthcare professionals involved in the treatment of EB may include general practitioners (GP), neonatologists, paediatricians, geneticists, dermatologists, specialists EB nurses, gastroenterologists, pain specialists, endocrinologists, oncologists, urologists, otolaryngologists (ear, nose and throat or ENT specialists), cardiologists, plastic surgeons, ophthalmologists, dentists, podiatrists, dieticians, psychologists, occupational therapists, physiotherapists, and orthotics.2,4 The need for different healthcare professionals may change over a person’s lifetime and extend beyond those listed here.
Please visit DEBRA Australia: EB Nurse Program for information on EB nurses in specific state hospitals.
Clinical care for rare diseases often involves a multidisciplinary team of medical, care and support professionals. Please note that the information provided here is as a guide and that RVA does not necessarily monitor or endorse specific clinics or health experts.
This may not be applicable to all rare diseases but for many, palliative care services may be relevant and useful. Palliative care services are available for people (adults, children and their families) living with a life-limiting illness and is not only for end-of-life care. It can also help at any stage of illness from diagnosis onwards, and will look different for different people. Palliative care services provide assistance, support, resources and tools to help people manage their illness and the symptoms, ease pain, and improve comfort and quality of life. If this is relevant to you and you wish to find out more information about palliative care and how it can help you, please visit:
")
Clinical Care Guidelines
DEBRA International is undertaking a long-term initiative to develop clinical practice guidelines (CPGs) for epidermolysis bullosa (EB), which provide recommendations for clinical care based on evidence gained from medical science and, when no evidence exists, on expert opinion.1 Please visit DEBRA Australia’s page on DEBRA International Clinical Practice Guidelines.
Emergency Management
Individuals living with rare diseases may have complex medical issues and disabilities, which are not always visible. It is often useful to refer to their medical history as well as personal information such as a medical card, doctor’s letter, or if available, a rare disease passport, for relevant information.
In addition, individuals, their parents, families and carers often develop extensive expertise on their specific rare disease. It is important to recognise that they can contribute valuable knowledge about their rare condition. Rare diseases often impact individuals differently, so it’s important to consider a person’s lived experience.
It may be important to consider the following when managing individuals living with EB at emergency departments/services:
Special care is required when treating and handling EB individuals who have fragile skin that can injure easily. DEBRA Australia’s page on DEBRA International Clinical Practice Guidelines includes clinical care recommendations, such as for skin and wound care, foot care, pain management and others.
Below are some emergency care recommendations from other countries that may be relevant or useful:
Research
Information on EB research in Australia can be found at:
There are specific considerations around participating in rare disease research, including clinical trials. It is important to be mindful of issues such as data privacy, research ethics, consent and differences in research regulations between Australia and other countries. For more information, please visit the RARE Portal’s Considerations for Participating in Health and Medical Research page.
If you are interested in finding clinical trials for your condition, please visit the following websites; however, there may not be any clinical trials available:
It is best to discuss your interest in research, including clinical trials, with your medical team to determine suitability and eligibility.
Rare Disease Organisation(s)
Australian Organisations:
DEBRA Australia
Website: https://www.debra.org.au/
DEBRA Australia provides support to those living with EB, including essential services such as EB Nurse and Family Support Program, medical supplies, aids and equipment, and EB research.
EB Research Partnerships Australia
Website: https://ebresearch.org.au/
EB Research Partnership Australia funds research aimed at treating and ultimately curing EB.
Cure EB
Website: https://cureeb.org.au/
Cure EB’s mission is to fund ground-breaking research, accelerate treatments, and bring hope to those living with EB. They are dedicated to driving scientific advancements that will lead to a cure.
Please note that RVA does not monitor or endorse each group/organisation’s operational governance and activities. When engaging with a group, please consider the information on the RARE Portal’s Finding Helpful Peer and Community Supports page.
Lived Experience
EB varies between individuals, and each person’s experience is unique.
Please visit DEBRA Australia: Our Stories and EB Research Partnership Australia: Life with EB Disease to read the personal stories of people living with EB.
Personal stories shared with Rare Voices Australia:
If you would like to share your personal story with RVA, please visit the Rare Voices Australia: Share Your Story page. RVA will consider your story for publishing on our website and inclusion on the RARE Portal.
Support Services and Resources
- Independence Australia: National Epidermolysis Bullosa Dressing Scheme – supports people with EB by providing affordable access to specialised bandages and dressings
- Australian Government Department of Health and Aged Care: National Epidermolysis Bullosa Dressing Scheme administrator – provides help with the costs of EB treatment
- DEBRA Australia: Family Support Program
- DEBRA Australia: Butterfly Breaks
- DEBRA Australia: EB Services
- EB Research Partnership Australia: EB Resources
For information on available government and social services that provide support for individuals with a rare disease, please visit the National and State Services pages.
Mental Health
People living with a rare disease often face unique challenges such as diagnostic delays, misdiagnoses, limited treatment options, and limited access to rare disease specialists and support. These challenges may impact people’s emotional wellbeing and quality of life. Many find it helpful to seek mental health and wellbeing support to cope with ongoing stress and uncertainty. Connecting with people who have shared experiences through a support group may also be helpful. Information about relevant mental health and wellbeing support can be found at:
- Mental Health and Wellbeing Support for Australians Living with a Rare Disease
- The National and State Services pages underneath the ‘Mental Health’ sections listed
DEBRA Australia’s Professional Counselling program can assist families to access professional counselling with psychologists who have experience in managing chronic illness and have had some background training about EB and its impact on individuals and families.
Other Information
Further information on EB can be found at:
- The Royal Children’s Hospital Melbourne: Epidermolysis bullosa
- The Sydney Children’s Hospitals Network: Epidermolysis bullosa handbook
- Children’s Health Queensland: Epidermolysis bullosa
- Genetic and Rare Diseases (GARD) Information Center: Epidermolysis bullosa
- National Organization for Rare Disorders (NORD): Epidermolysis Bullosa
- EB Research Partnership (United States of America)
Useful Links for Healthcare Professionals
DEBRA Australia: Health Professionals
Australasian College of Dermatologists (ACD): Epidermolysis Bullosa
References
- The Australasian College of Dermatologists (ACD). A to Z of skin: Epidermolysis Bullosa. 2019. Updated April 2023. Accessed 7 November 2024. https://www.dermcoll.edu.au/atoz/epidermolysis-bullosa/
- The Royal Children’s Hospital Melbourne. Epidermolysis bullosa. Reviewed August 2020. Accessed 7 November 2024. https://www.rch.org.au/kidsinfo/fact_sheets/Epidermolysis_bullosa/
- National Organization for Rare Disorders (NORD). Epidermolysis Bullosa. 2013. Updated 22 January 2024. Accessed 7 November 2024. https://rarediseases.org/rare-diseases/epidermolysis-bullosa/
- Khanna D, Bardhan A. Epidermolysis Bullosa. Updated 11 January 2024. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Accessed 13 January 2025. https://www.ncbi.nlm.nih.gov/books/NBK599531/
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat. Rev. Dis. Primers. 2020; 6:78. https://doi.org/10.1038/s41572-020-0210-0
- Genetic and Rare Diseases (GARD) Information Center. Epidermolysis bullosa. Accessed 7 November 2024. https://rarediseases.info.nih.gov/diseases/6359/epidermolysis-bullosa
Contributors
This page has been co-developed by Rare Voices Australia (RVA)’s RARE Portal team in consultation with DEBRA Australia and EB Research Partnership Australia.
If you are aware of any additional information that may benefit stakeholders with an interest in this page, or if you notice any broken links or inaccurate information, please let us know via the Contribute page.
